distToNearest - Distance to nearest neighbor
Description¶
Get non-zero distance of every heavy chain (IGH) sequence (as defined by
sequenceColumn) to its nearest sequence in a partition of heavy chains sharing the same
V gene, J gene, and junction length (V-J-length), or in a partition of single cells with heavy/long chains
sharing the same heavy/long chain V-J-length combination, or of single cells with heavy/long and light/short chains
sharing the same heavy/long chain V-J-length and light/short chain V-J-length combinations.
Usage¶
distToNearest(
db,
sequenceColumn = "junction",
vCallColumn = "v_call",
jCallColumn = "j_call",
model = c("ham", "aa", "hh_s1f", "hh_s5f", "mk_rs1nf", "mk_rs5nf", "m1n_compat",
"hs1f_compat"),
normalize = c("len", "none"),
symmetry = c("avg", "min"),
first = TRUE,
VJthenLen = TRUE,
nproc = 1,
fields = NULL,
cross = NULL,
mst = FALSE,
subsample = NULL,
progress = FALSE,
cellIdColumn = NULL,
locusColumn = "locus",
locusValues = c("IGH"),
onlyHeavy = TRUE,
keepVJLgroup = TRUE
)
Arguments¶
- db
- data.frame containing sequence data.
- sequenceColumn
- name of the column containing the junction for grouping and for calculating nearest neighbor distances. Note that while both heavy/long and light/short chain junctions may be used for V-J-length grouping, only the heavy/long chain (IGH, TRB, TRD) junction is used to calculate distances.
- vCallColumn
- name of the column containing the V-segment allele calls.
- jCallColumn
- name of the column containing the J-segment allele calls.
- model
- underlying SHM model, which must be one of
c("ham", "aa", "hh_s1f", "hh_s5f", "mk_rs1nf", "hs1f_compat", "m1n_compat"). See Details for further information. - normalize
- method of normalization. The default is
"len", which divides the distance by the length of the sequence group. If"none"then no normalization if performed. - symmetry
- if model is hs5f, distance between seq1 and seq2 is either the average (avg) of seq1->seq2 and seq2->seq1 or the minimum (min).
- first
- if
TRUEonly the first call of the gene assignments is used. ifFALSEthe union of ambiguous gene assignments is used to group all sequences with any overlapping gene calls. - VJthenLen
- logical value specifying whether to perform partitioning as a 2-stage
process. If
TRUE, partitions are made first based on V and J gene, and then further split based on junction lengths corresponding tosequenceColumn. IfFALSE, perform partition as a 1-stage process during which V gene, J gene, and junction length are used to create partitions simultaneously. Defaults toTRUE. - nproc
- number of cores to distribute the function over.
- fields
- additional fields to use for grouping.
- cross
- character vector of column names to use for grouping to calculate distances across groups. Meaning the columns that define self versus others.
- mst
- if
TRUE, return comma-separated branch lengths from minimum spanning tree. - subsample
- number of sequences to subsample for speeding up pairwise-distance-matrix calculation.
Subsampling is performed without replacement in each V-J-length group of heavy chain sequences.
If
subsampleis larger than the unique number of heavy chain sequences in each VJL group, then the subsampling process is ignored for that group. For each heavy chain sequence indb, the reporteddist_nearestis the distance to the closest heavy chain sequence in the subsampled set for the V-J-length group. IfNULLno subsampling is performed. - progress
- if
TRUEprint a progress bar. - cellIdColumn
- name of the character column containing cell identifiers or barcodes.
If specified, grouping will be performed in single-cell mode
with the behavior governed by the
locusColumnandonlyHeavyarguments. If set toNULLthen the bulk sequencing data is assumed. - locusColumn
- name of the column containing locus information. Valid loci values are “IGH”, “IGI”, “IGK”, “IGL”, “TRA”, “TRB”, “TRD”, and “TRG”.
- locusValues
- Loci values to focus the analysis on.
- onlyHeavy
- use only the IGH (BCR) or TRB/TRD (TCR) sequences
for grouping. Only applicable to single-cell data.
Ignored if
cellIdColumn=NULL. See groupGenes for further details. - keepVJLgroup
- logical value specifying whether to keep in the output the the column
column indicating grouping based on V-J-length combinations. Only applicable for
1-stage partitioning (i.e.
VJthenLen=FALSE). Also see groupGenes.
Value¶
Returns a modified db data.frame with nearest neighbor distances between heavy chain
sequences in the dist_nearest column if cross=NULL. If cross was
specified, distances will be added as the cross_dist_nearest column.
Note that distances between light/short (IGK, IGL, TRA, TRG) chain sequences are not calculated,
even if light/short chains were used for V-J-length grouping via onlyHeavy=FALSE.
Light/short chain sequences, if any, will have NA in the dist_nearest output column.
Note that the output vCallColumn and jCallColumn columns will be converted to
type character if they were type factor in the input db.
Details¶
To invoke single-cell mode the cellIdColumn argument must be specified and locusColumn
must be correct. Otherwise, distToNearest will be run with bulk sequencing assumptions,
using all input sequences regardless of the values in the locusColumn column.
Under single-cell mode, only heavy/long chain (IGH, TRB, TRD) sequences will be used for calculating
nearest neighbor distances. Under non-single-cell mode, all input sequences will be used for
calculating nearest neighbor distances, regardless of the values in the locusColumn field (if present).
Values in the locusColumn must be one of c("IGH", "IGI", "IGK", "IGL") for BCR
or c("TRA", "TRB", "TRD", "TRG") for TCR sequences. Otherwise, the function returns an
error message and stops.
For single-cell mode, the input format is the same as that for groupGenes.
Namely, each row represents a sequence/chain. Sequences/chains from the same cell are linked
by a cell ID in the cellIdColumn field. In this mode, there is a choice of whether
grouping should be done by (a) using IGH (BCR) or TRB/TRD (TCR) sequences only or
(b) using IGH plus IGK/IGL (BCR) or TRB/TRD plus TRA/TRG (TCR).
This is governed by the onlyHeavy argument.
Note, distToNearest required that each cell (each unique value in cellIdColumn)
correspond to only a single IGH (BCR) or TRB/TRD (TCR) sequence.
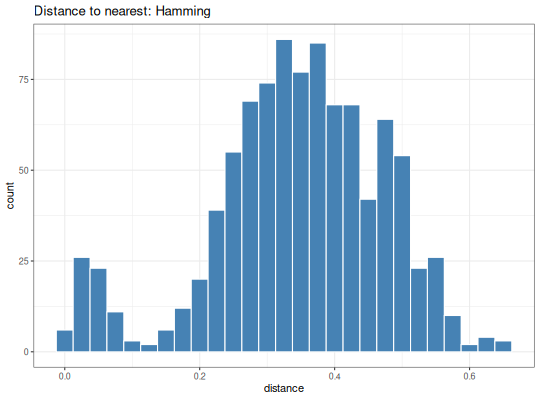
The distance to nearest neighbor can be used to estimate a threshold for assigning Ig sequences to clonal groups. A histogram of the resulting vector is often bimodal, with the ideal threshold being a value that separates the two modes.
The following distance measures are accepted by the model parameter.
"ham": Single nucleotide Hamming distance matrix from getDNAMatrix with gaps assigned zero distance."aa": Single amino acid Hamming distance matrix from getAAMatrix."hh_s1f": Human single nucleotide distance matrix derived from HH_S1F with calcTargetingDistance."hh_s5f": Human 5-mer nucleotide context distance matix derived from HH_S5F with calcTargetingDistance."mk_rs1nf": Mouse single nucleotide distance matrix derived from MK_RS1NF with calcTargetingDistance."mk_rs5nf": Mouse 5-mer nucleotide context distance matrix derived from MK_RS1NF with calcTargetingDistance."hs1f_compat": Backwards compatible human single nucleotide distance matrix used in SHazaM v0.1.4 and Change-O v0.3.3."m1n_compat": Backwards compatibley mouse single nucleotide distance matrix used in SHazaM v0.1.4 and Change-O v0.3.3.
Note on NAs: if, for a given combination of V gene, J gene, and junction length,
there is only 1 heavy chain sequence (as defined by sequenceColumn), NA is
returned instead of a distance (since it has no heavy/long chain neighbor). If for a given combination
there are multiple heavy/long chain sequences but only 1 unique one, (in which case every heavy/long cahin
sequence in this group is the de facto nearest neighbor to each other, thus giving rise to distances
of 0), NAs are returned instead of zero-distances.
Note on subsample: Subsampling is performed independently in each V-J-length group for
heavy/long chain sequences. If subsample is larger than number of heavy/long chain sequences
in the group, it is ignored. In other words, subsampling is performed only on groups in which the
number of heavy/long chain sequences is equal to or greater than subsample. dist_nearest
has values calculated using all heavy chain sequences in the group for groups with fewer than
subsample heavy/long chain sequences, and values calculated using a subset of heavy/long chain
sequences for the larger groups. To select a value of subsample, it can be useful to explore
the group sizes in db (and the number of heavy/long chain sequences in those groups).
References¶
- Smith DS, et al. Di- and trinucleotide target preferences of somatic mutagenesis in normal and autoreactive B cells. J Immunol. 1996 156:2642-52.
- Glanville J, Kuo TC, von Budingen H-C, et al. Naive antibody gene-segment frequencies are heritable and unaltered by chronic lymphocyte ablation. Proc Natl Acad Sci USA. 2011 108(50):20066-71.
- Yaari G, et al. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front Immunol. 2013 4:358.
Examples¶
# Subset example data to one sample as a demo
data(ExampleDb, package="alakazam")
db <- subset(ExampleDb, sample_id == "-1h")
# Use genotyped V assignments, Hamming distance, and normalize by junction length
# First partition based on V and J assignments, then by junction length
# Take into consideration ambiguous V and J annotations
dist <- distToNearest(db, sequenceColumn="junction",
vCallColumn="v_call_genotyped", jCallColumn="j_call",
model="ham", first=FALSE, VJthenLen=TRUE, normalize="len")
# Plot histogram of non-NA distances
p1 <- ggplot(data=subset(dist, !is.na(dist_nearest))) +
theme_bw() +
ggtitle("Distance to nearest: Hamming") +
xlab("distance") +
geom_histogram(aes(x=dist_nearest), binwidth=0.025,
fill="steelblue", color="white")
plot(p1)
See also¶
See calcTargetingDistance for generating nucleotide distance matrices from a TargetingModel object. See HH_S5F, HH_S1F, MK_RS1NF, getDNAMatrix, and getAAMatrix for individual model details. getLocus to get locus values based on allele calls.