findThreshold - Find distance threshold
Description¶
findThreshold automatically determines an optimal threshold for clonal assignment of
Ig sequences using a vector of nearest neighbor distances. It provides two alternative methods
using either a Gamma/Gaussian Mixture Model fit (method="gmm") or kernel density
fit (method="density").
Usage¶
findThreshold(
distances,
method = c("density", "gmm"),
edge = 0.9,
cross = NULL,
subsample = NULL,
model = c("gamma-gamma", "gamma-norm", "norm-gamma", "norm-norm"),
cutoff = c("optimal", "intersect", "user"),
sen = NULL,
spc = NULL,
progress = FALSE
)
Arguments¶
- distances
- numeric vector containing nearest neighbor distances.
- method
- string defining the method to use for determining the optimal threshold.
One of
"gmm"or"density". See Details for methodological descriptions. - edge
- upper range as a fraction of the data density to rule initialization of
Gaussian fit parameters. Default value is 90
Applies only when
method="density". . - cross
- supplementary nearest neighbor distance vector output from distToNearest
for initialization of the Gaussian fit parameters.
Applies only when
method="gmm". - subsample
- maximum number of distances to subsample to before threshold detection.
- model
- allows the user to choose among four possible combinations of fitting curves:
"norm-norm","norm-gamma","gamma-norm", and"gamma-gamma". Applies only whenmethod="gmm". - cutoff
- method to use for threshold selection: the optimal threshold
"opt", the intersection point of the two fitted curves"intersect", or a value defined by user for one of the sensitivity or specificity"user". Applies only whenmethod="gmm". - sen
- sensitivity required. Applies only when
method="gmm"andcutoff="user". - spc
- specificity required. Applies only when
method="gmm"andcutoff="user". - progress
- if
TRUEprint a progress bar.
Value¶
"gmm"method: Returns a GmmThreshold object including the
thresholdand the function fit parameters, i.e. mixing weight, mean, and standard deviation of a Normal distribution, or mixing weight, shape and scale of a Gamma distribution."density"method: Returns a DensityThreshold object including the optimumthresholdand the density fit parameters.
Details¶
"gmm": Performs a maximum-likelihood fitting procedure, for learning the parameters of two mixture univariate, either Gamma or Gaussian, distributions which fit the bimodal distribution entries. Retrieving the fit parameters, it then calculates the optimum thresholdmethod="optimal", where the average of the sensitivity plus specificity reaches its maximum. In addition, thefindThresholdfunction is also able to calculate the intersection point (method="intersect") of the two fitted curves and allows the user to invoke its value as the cut-off point, instead of optimal point."density": Fits a binned approximation to the ordinary kernel density estimate to the nearest neighbor distances after determining the optimal bandwidth for the density estimate via least-squares cross-validation of the 4th derivative of the kernel density estimator. The optimal threshold is set as the minimum value in the valley in the density estimate between the two modes of the distribution.
Note¶
Visually inspecting the resulting distribution fits is strongly recommended when using either fitting method. Empirical observations imply that the bimodality of the distance-to-nearest distribution is detectable for a minimum of 1,000 distances. Larger numbers of distances will improve the fitting procedure, although this can come at the expense of higher computational demands.
Examples¶
# Subset example data to 50 sequences, one sample and isotype as a demo
data(ExampleDb, package="alakazam")
db <- subset(ExampleDb, sample_id == "-1h" & c_call=="IGHG")[1:50,]
# Use nucleotide Hamming distance and normalize by junction length
db <- distToNearest(db, sequenceColumn="junction", vCallColumn="v_call",
jCallColumn="j_call", model="ham", normalize="len", nproc=1)
# Find threshold using the "gmm" method with user defined specificity
output <- findThreshold(db$dist_nearest, method="gmm", model="gamma-gamma",
cutoff="user", spc=0.99)
plot(output, binwidth=0.02, title=paste0(output@model, " loglk=", output@loglk))
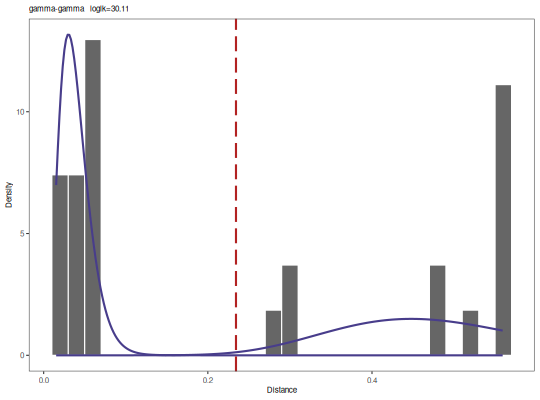
print(output)
[1] 0.2342911
See also¶
See distToNearest for generating the nearest neighbor distance vectors. See plotGmmThreshold and plotDensityThreshold for plotting output.