SHM targeting models¶
The targeting model is the background likelihood of a particular mutation, based on the surrounding sequence context as well as the mutation itself. The model is inferred from observed mutations in the data. The model can then be transformed into a distance function to compare Ig sequences of a given dataset based on the likelihood of the observed mutations. This is done via the following steps:
- Infer a substitution model, which is the likelihood of a base mutating to each other base given the microsequence context.
- Infer a mutability model, which is likelihood of a given base being mutated given the microsequence context and substitution model.
- Visualize the mutability model to identify hot and cold spots.
- Calculate a nucleotide distance matrix based on the underlying SHM models.
Example data¶
A small example AIRR Rearrangement database is included in the alakazam package.
Inferring a targeting model requires the following fields (columns) to
be present in the table:
sequence_idsequence_alignmentgermline_alignment_d_maskv_call
# Import required packages
library(shazam)
# Load example data
data(ExampleDb, package="alakazam")
# Subset to IGHG for faster usage demonstration
db <- subset(ExampleDb, c_call == "IGHG")
Infer targeting model (substitution and mutability)¶
The function for inferring substitution rates (createSubstitutionMatrix)
counts the number of mutations from a given base to all others occurring in the
center position for all 5-mer motifs in the dataset. The model argument of
createSubstitutionMatrix allows the user to specify whether to count all
mutations, or just silent mutations to infer the model. Column names for the
sample sequence, germline sequence, and V call can also be passed in as
parameters if they differ from Change-O defaults. Additionally, the
multipleMutation parameter determines handling of multiple mutations in a
single 5-mer: independent treats each mutation independently and ignore
entirely disregards 5-mers with multiple mutations.
# Create substitution model using silent mutations
sub_model <- createSubstitutionMatrix(db, model="s",
sequenceColumn="sequence_alignment",
germlineColumn="germline_alignment_d_mask",
vCallColumn="v_call")
The function for inferring a mutability model (createMutabilityMatrix) counts
the number of mutations in all 5-mer motifs of the dataset, and depends upon the
inferred substitution rates. Similar parameters as those available for
inferring the substitution rates are available to adjust this function.
# Create mutability model using silent mutations
mut_model <- createMutabilityMatrix(db, sub_model, model="s",
sequenceColumn="sequence_alignment",
germlineColumn="germline_alignment_d_mask",
vCallColumn="v_call")
createMutabilityMatrix creates an object of class MutabilityModel that contains
a named numeric vector of 1024 normalized mutability. The numbers of silent and
replacement mutations used for estimating the 5-mer mutabilities are recorded in
the numMutS and numMutR slots, respectively. rates. The source slot contains
a named vector indicating whether each 5-mer mutability was inferred or measured.
A data.frame with both the mutability values and derivation source.
# Number of silent mutations used for estimating 5-mer mutabilities
mut_model@numMutS
# Number of replacement mutations used for estimating 5-mer mutabilities
mut_model@numMutR
# Mutability and source as a data.frame
head(as.data.frame(mut_model))
The inferred substitution and mutability models returned by the above functions
only account for unambiguous 5-mers. However, there may be cases in which the
user may need the likelihood of a mutation in a 5-mer with ambiguous characters.
Each of the above functions has a corresponding function (extendSubstitutionMatrix
and extendMutabilityMatrix) to extend the models to infer 5-mers with Ns by
averaging over all corresponding unambiguous 5-mers.
# Extend models to include ambiguous 5-mers
sub_model <- extendSubstitutionMatrix(sub_model)
mut_model <- extendMutabilityMatrix(mut_model)
These extended substitution and mutability models can be used to create an
overall SHM targeting matrix (createTargetingMatrix), which is the combined
probability of mutability and substitution.
# Create targeting model matrix from substitution and mutability models
tar_matrix <- createTargetingMatrix(sub_model, mut_model)
All of the above steps can be combined by using the single function
createTargetingModel to infer a TargetingModel object directly from
the dataset. Again, the numbers of silent and replacement mutations used
for estimating the 5-mer mutabilities are also recorded in the numMutS and
numMutR slots respectively.
Additionally, it is generally appropriate to consider the mutations
within a clone only once. Consensus sequences for each clone can be generated
using the collapseClones function.
# Collapse sequences into clonal consensus
clone_db <- collapseClones(db, cloneColumn="clone_id",
sequenceColumn="sequence_alignment",
germlineColumn="germline_alignment_d_mask",
nproc=1)
# Create targeting model in one step using only silent mutations
# Use consensus sequence input and germline columns
model <- createTargetingModel(clone_db, model="s", sequenceColumn="clonal_sequence",
germlineColumn="clonal_germline", vCallColumn="v_call")
Visualize targeting model¶
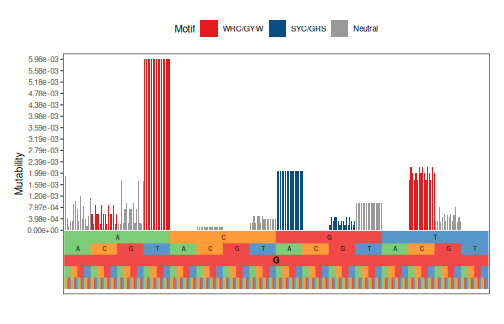
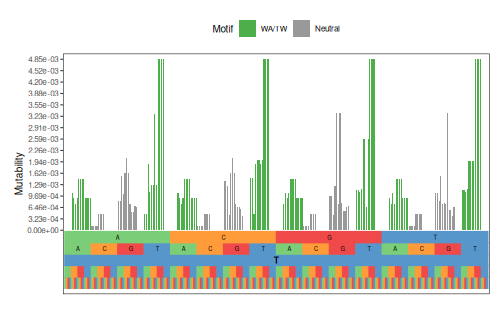
The visualization of a dataset’s underlying SHM mutability model can be used to
investigate hot and cold spot motifs. The length of the bars on the plot of
mutability rates corresponds to the likelihood of a given base in the given
5-mer being mutated. The plotting function plotMutability has an argument
style to specify either a hedgehog plot (circlular) or barplot diplay of
5-mer mutability rates. If the mutability for only specific bases is required,
this can be specified via the nucleotides argument.
# Generate hedgehog plot of mutability model
plotMutability(model, nucleotides="A", style="hedgehog")

plotMutability(model, nucleotides="C", style="hedgehog")

# Generate bar plot of mutability model
plotMutability(model, nucleotides="G", style="bar")
plotMutability(model, nucleotides="T", style="bar")
Calculate targeting distance matrix¶
In the Change-O pipeline, the hs5f cloning method rely on an inferred
targeting model. If users wish to use a targeting model inferred from their
data to assign distance between sequences for clonal grouping, then the observed
SHM targeting rates must be transformed into distances. The
calcTargetingDistance function returns a matrix of distances between each 5-mer
and each corresponding mutation of the center base. This matrix can also be
generated and written directly to a file using the function
writeTargetingDistance.
# Calculate distance matrix
dist <- calcTargetingDistance(model)